- 移动端


上海吉凯基因医学科技股份有限公司品牌商
15 年
手机商铺
- NaN
- 0.5
- 0.5
- 1.5
- 0.5





公司新闻/正文
《Circulation》: 杜杰教授课题组揭示心肌缺血再灌注损伤新机制
7830 人阅读发布时间:2019-09-23 14:05
心肌细胞缺血再灌注损伤作为一个重要的临床问题至今没有有效的治疗手段。虽然普遍认为线粒体功能异常、氧化应激及炎症反应是缺血再灌注损伤过程的主要分子机制,但具体机制目前尚不清楚。
2019年6月21日首都医科大学附属北京安贞医院杜杰教授课题组在《Circulation》(影响因子23.054)上在线发表了题为“S100a8/a9 Signaling Causes Mitochondrial Dysfunction and Cardiomyocytes Death in Response Ischemic/Reperfusion Injury” 的文章揭示了缺血再灌注损伤中心肌细胞死亡的分子机制。

该研究中作者利用动态转录组分析,发现心肌缺血再灌注(MI/R)损伤的早期关键分子S100a8/a9。直接证明了S100a8/a9通过抑制TLR4/Erk 调节的PGC-1/NRF1信号导致线粒体功能异常并导致线粒体复合体Ⅰ抑制。揭示了炎症分子与线粒体功能异常间的新联系以及MI/R损伤的潜在药物治疗靶点。
首先作者通过转录组学分析(图1. A、B、C)发现在MI/R损伤中S100a8/a9是早期阶段上调最明显的基因(图1. D、E)。
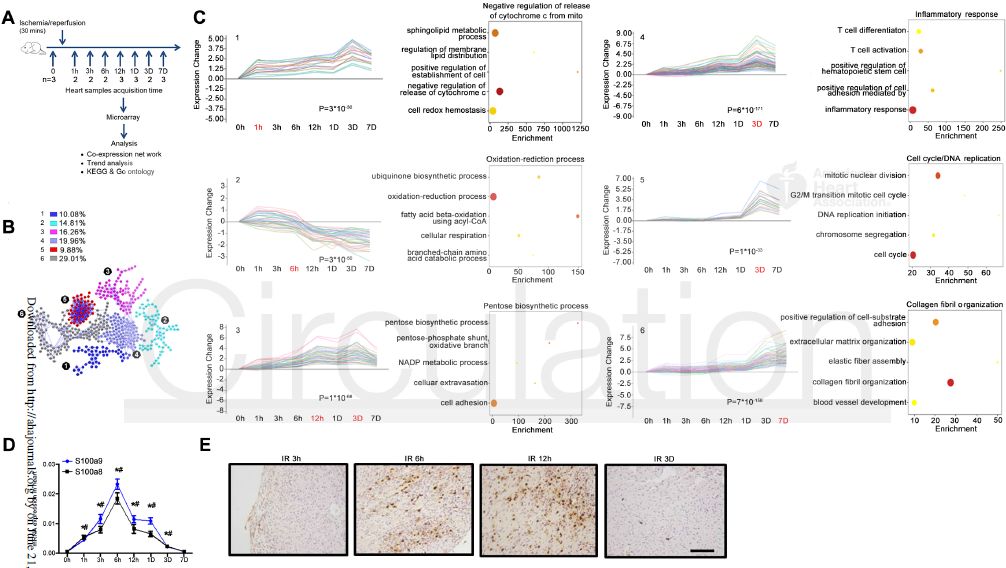
图1. MI/R损伤中S100a8/a9 动态变化
那么S100a8/a9扮演着什么角色呢?本研究发现在MI/R损伤中S100a8/a9促进心肌细胞死亡。作者利用S100a9敲除小鼠和S100a9过表达小鼠构建MI/R损伤模型,较之对照组,发现S100a9敲除小鼠的心肌梗死面积明显变小、心脏收缩功能提高、心肌细胞死亡显著减少,并且在造模后28时心脏收缩功能异常及心肌纤维化的症状消失,而S100a9过表达小鼠则相反,表型显著恶化(图2)。

图2. MI/R损伤中S100a8/a9促进心肌细胞死亡和心衰
接着作者通过转录组分析(图3. A),发现I/R一天后S100a9过表达小鼠心脏及重组S100a8/a9处理过的小鼠新生心肌细胞中上调的基因主要集中于炎症相关信号(图3. B,C),这与已知的S100a8/a9 扮演促炎角色相一致。此外还发现主要明显下调的通路为线粒体氧化应激,且大部分下调的差异表达基因属于线粒体电子传递链(ETC)复合体Ⅰ的亚基(图3. G-P)。基于此作者将关注点聚焦在了S100a8/a9与线粒体复合体Ⅰ上,使用吉凯基因提供的腺病毒产品(Ad-NUDFS7和Ad-control)在新生心肌细胞中过表达NUDFS7突变体,发现S100a8/a9通过抑制NUDFs基因转录损伤线粒体复合体Ⅰ激活引起心肌细胞死亡。

图3. S100a8/a9抑制ETC复合体Ⅰ和呼吸功能
研究表明NRF1直接结合大多数NUDFs基因的启动子,PGC-1α与NRF1共激活调控基因表达是维持线粒体内环境稳态必不可少的。作者发现S100a8/a9通过TLR4-Erk信号抑制PGC-1α/NRF1下调NDUFs基因转录(图4. K)。

图4. S100a8/a9通过TLR4/Erk信号通路抑制PGC-1α/NRF-1
接下来作者猜想通过阻遏S100a8/a9可能预防MI/R损伤。于是作者构建了S100a9中和抗体,研究发现S100a9中和抗体处理后小鼠MI/R损伤显著减轻。此外还发现MI/R损伤发生后CXCL1募集CXCR2 阳性细胞分泌S100a8/a9。进一步对S100a8/a9升高与MI/R损伤之间的临床相关性予以分析,发现心肌梗死患者血清内S100a8/a9水平显著高于健康对照组人群,且发现经皮冠状动脉治疗后一天患者血清内S100a8/a9 水平与后期心源性休克、心衰等发生风险显著相关(图5)。
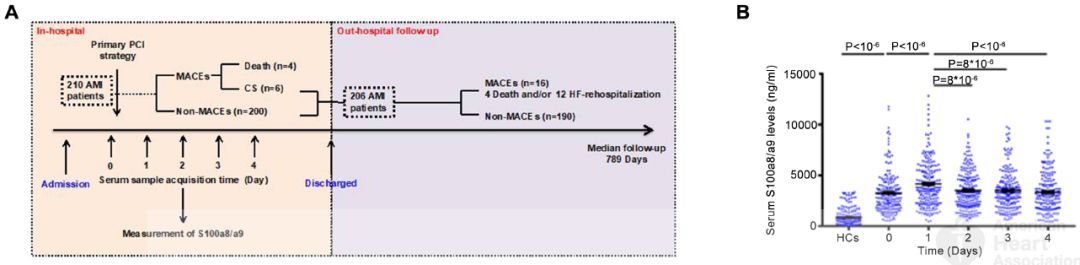
图5. 血清中S100a8/a9水平检测
总结
1. S100a8/a9 加重缺血再灌注损伤,主要影响线粒体功能异常(图6);
2. S100a8/a9是一个新的预后情况分子标志物;
3. S100a8/a9有望成为治疗心肌缺血再灌注损伤的新靶点。

图6. I/R模型中S100a8/a9作用机制
吉凯助力
该研究中腺病毒产品:Ad-NDUFS7及对照病毒由吉凯基因提供。通过感染新生心肌细(NCMs),使其有效的过表达NDUFS7(图7),进而帮助证明了S100a8/a9是通过抑制NDFUs基因的转录导致心肌细胞死亡。
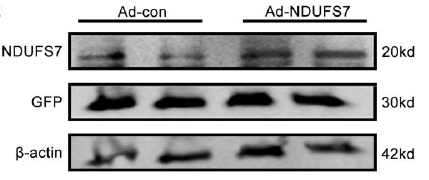
图7. Western blot检测NDUFS7过表达效果
作者简介
首都医科大学附属北京安贞医院杜杰教授、李玉琳副教授和美国托马斯杰斐逊大学马新亮教授为本文的通讯作者,首都医科大学附属北京安贞医院李玉琳副教授为本文第一作者。
