- 移动端


上海吉凯基因医学科技股份有限公司品牌商
15 年
手机商铺
- NaN
- 0.5
- 0.5
- 1.5
- 0.5





公司新闻/正文
蛋白对接工具怎么选?从传统对接方式到深度学习方法,收藏这篇就够了!
1458 人阅读发布时间:2025-11-10 17:16
蛋白-蛋白相互作用对许多生物活动至关重要,这些相互作用的结构特征可以阐明蛋白质在生命系统中的功能,以及设计具有高特异性和低毒性的新疗法。
伊朗拉兹大学的Mina Barhoon和Hamid Mahdiuni总结了现有蛋白-蛋白对接的工具以及他们的优缺点,方便研究者根据课题需要选用合适的蛋白对接工具。
本文整理了这篇综述的精华内容,一次性说清蛋白对接工具的选择策略,强烈建议收藏!

PubMed报道的近些年蛋白-蛋白对接工具使用趋势
01 蛋白对接策略
✦ 搜索算法用于探索蛋白质的构象空间并生成可能的结合构象
✦ 打分函数则基于物理、化学和统计模型对这些构象进行评价和排序
搜索算法的设计至关重要:一方面需确保真实结合构象不被漏选,另一方面也应具备从大量候选构象中准确识别最优构象的能力。
不同的对接工具在搜索策略上各有特点,可大致按全局/局部、基于模板/从头预测、刚性/柔性等维度进行分类。

蛋白-蛋白对接搜索算法比较
局部搜索 vs 全局搜索
局部搜索算法需要预先定义对接位点,探索有限刚性构象空间或者局部侧链柔性构象。这种算法节约计算资源,但是假定位点需要先验知识并且只能探索有限的构象。
全局搜索算法搜索全部蛋白表面,不需要指定位点,依赖对蛋白的转动和平移自由度进行大量采样,需消耗大量计算资源。全局搜索适合发现新的结合方式,而局部搜索可以提高已知结合位点的蛋白对接精度。
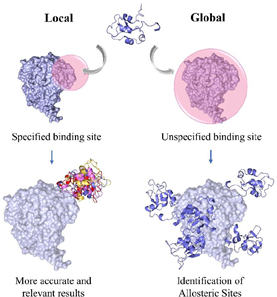
局部搜索和全局搜索算法比较
基于模板 vs 从头算
基于模板算法和从头算代表预测蛋白复合物的不同路径。
基于模板对接利用了结构同源性,首先需要确定一个合适的模板(通常序列相似度>30%)。例如HADDOCK使用两两比对将目标序列与模板序列进行比较,然后进行同源建模,并在界面约束(如氢键、距离等)的引导下进行受约束的分子动力学模拟。HADDOCK的最/新更新整合了生化数据和改进的评分函数,从而增强了基于模板的预测能力。 这种方法假定存在稳定的相互作用模式,因此在有高质量模板可用的情况下效率较高。
从头算直接预测新的复合物结构,其依据的是物理化学原理,无需模板。
它采用了诸如遗传算法或蒙特卡罗等采样技术,如在RosettaDock中所见。使用粗粒度模型来生成数千种构象,然后通过基于物理原理的能量函数进行全原子优化。
AlphaFold-Multimer使用深度学习的方法提高了从头算的预测能力,并且减少了构象空间采样数量同时提高了计算精度。
这种算法能够识别接近天然复合物的构象,但是由于缺乏结构限制仍然消耗大量计算资源。
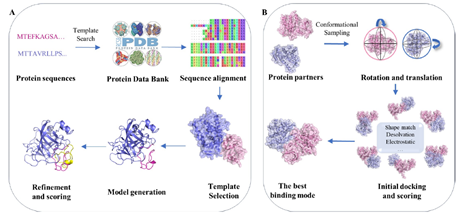
两种预测蛋白质复合物算法
A基于模版算法;B从头算
刚性对接 vs 柔性对接
刚性对接与柔性对接是蛋白对接中两种主要策略。蛋白质结合过程中常发生显著的构象变化,柔性对接成为该领域的关键挑战。
刚性对接方法(如 ClusPro 和 ZDOCK)通常仅优化配体与受体的位置和朝向,忽略构象柔性。相比之下,柔性对接更为复杂,可涉及从侧链调整到主链重排等多个层次的结构变化。
柔性对接通常采用两类策略:局部优化和全局构象调整。
例如,RosettaDock 通过蒙特卡洛方法(Monte Carlo)主要调整侧链和loop区域,而 AlphaRED 则通过能量最小化对关键残基进行优化。粗粒化模型(如 ResDock 和 UNRES-Dock)能够在较低计算成本下处理部分柔性,而 SwarmDock 利用模式分析、HADDOCK 借助分子动力学模拟处理全局性的大尺度构象重排。
ReplicaDock 通过复本交换策略增强构象采样效率,LATENTDOCK 则基于深度学习中的扩散模型捕捉完全柔性条件下的构象变化。
柔性对接虽然更准确地反映生物真实状态,但其计算速度通常较慢、资源消耗更大。
02 蛋白-蛋白对接的评分函数
蛋白-蛋白对接的评分函数在原理和方法上多种多样:
-
基于能量的方法:如ClusPro、RosettaDock等利用分子力学项进行评分。
-
基于知识的方法:如PatchDock和SymmDock,基于原子接触频率评分。
-
经验方法:如HawkDock结合MM/GBSA数据优化权重。
-
基于形状评分:如HDOCK和IDPdock,强调形状互补性。
-
深度学习工具:如ComDock的TRScore、GeoDock的损失函数。
-
共识方法:如InterEvDock3和FRODOCK,结合多种函数以提高可靠性。
不同的评分函数在计算速度、准确性和可靠性上具有不同优势,这些方法满足了从详细能量研究到复杂结构预测的多样化需求。
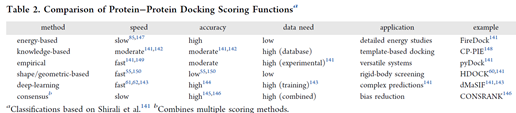
蛋白-蛋白对接评分函数比较
03 评价蛋白-蛋白对接结果的指标
通过多种指标评价蛋白-蛋白对接结果的准确性和可信度:
✦RMSD值: 比较预测结构和实验结构之间的几何偏差,值越小预测越准确.
✦天然接触分数FNat:评估在蛋白相互作用表面截断距离内正确接触残基的比率.
✦ DockQ:整合了FNat、LRMSD和IRMSD成为单一的评分.
✦ 对接成功率:计算排名前N个结果中正确结果的比率.
在CAPRI中, 预测模型被分为三个等级:
✦高质量:FNat ≥ 0.5, IRMSD ≤ 1.0 Å, LRMSD ≤ 1.0 Å, DockQ ≥ 0.80
✦中等质量:FNat ≥ 0.3, IRMSD ≤ 2.0Å, LRMSD ≤ 5.0 Å, DockQ 0.49−0.80
✦可接受的:FNat≥ 0.1, IRMSD ≤ 4.0 Å, LRMSD ≤ 10.0 Å, DockQ 0.23−0.49
04 传统蛋白-蛋白对接工具
Z-Dock
Z-DOCK是广泛应用的蛋白-蛋白对接软件,它采用刚性对接算法,基于快速傅立叶变换(FFT)高效遍历蛋白质分子之间所有可能的平移和旋转组合,生成大量的可能结合构象。打分函数结合了形状互补性、静电作用和去溶剂化能进行综合评价。
ZDOCK SCORE分数越高结合可能性越大,ZRANK SCORE分数越低结合越紧密。ZDOCK可通过聚类分析去除相似构象,也可以通过参数设置指定可能结合残基和排除不结合残基。
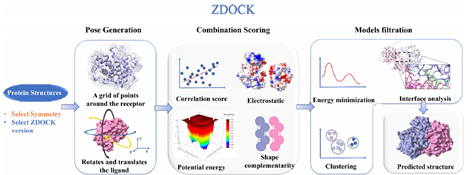
Z-DOCK工作流程
HADDOCK
HADDOCK利用来自实验或生物信息学提供的模糊相互作用信息来引导和约束对接过程。
HADDOCK的复合物结构预测过程分为三个阶段:
第一阶段根据用户提供的活性残基信息,HADDOCK生成相应的距离约束,并对配体分子进行随机旋转和平移。
该过程在模糊距离约束的引导下进行,不符合约束的构象会在能量评分中受到惩罚。随后对生成的初始复合物进行初步的能量最小化,在此过程中侧链可发生小幅移动,而主链保持刚性。此阶段将生成数千个可能的复合物构象,并基于HADDOCK评分进行初步排序和筛选,优选结构进入下一阶段。
第二阶段为半柔性模拟退火
HADDOCK自动将活性残基一定范围内的残基定义为界面区域,并在该区域内引入柔性,允许这些残基的侧链和主链发生移动。通过一个从高温到低温的模拟退火分子动力学过程,使界面区域在能量势场中松弛并优化构象。
第三阶段为显式溶剂精修
HADDOCK将上一步得到的优化结构置于显式水分子环境中,替代原先的隐式溶剂模型,以更接近真实生理条件。在该体系下进行短时间的分子动力学模拟和能量最小化,使水分子和离子在界面处重新排布,从而优化氢键和静电相互作用。最终计算每个结构的HADDOCK评分,并据此进行排名。该评分是一个加权综合评价指标,分值越低代表能量越优、结构越可靠。
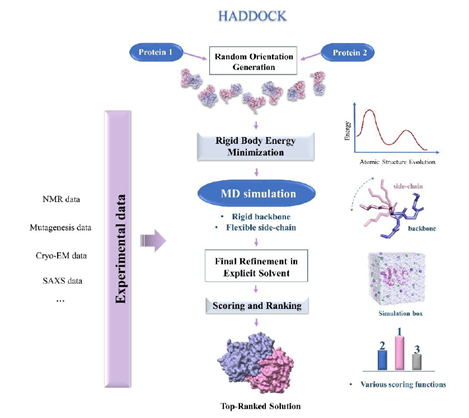
HADDOCK工作流程
应用建议:当用户具备任何形式的实验数据时,HADDOCK可作为首/选工具。
05 蛋白-蛋白对接深度学习工具
深度学习方法彻底改变了蛋白质-蛋白质对接的研究范式。与传统依赖构象采样和评分的策略不同,深度学习能够高效处理大规模蛋白质数据库,并显著提高了对接结构的预测精度。
该类方法在对接流程中扮演多种角色
例如:DockGPT 和 EquiDock 利用扩散模型作为搜索算法,探索构象空间并生成复合物结构;DeepRank-GNN 可对初始对接构象进行优化;而 DeepRank、PPI-Affinity 和 GeoDock 则用于评估复合物质量及预测结合亲和力。
此外,深度学习既适用于有模板场景(如 AlphaFold-Multimer和RoseTTAFold),也可处理无模板情景(如 D-I-TASSER),仅需输入序列信息即可完成预测。
深度学习方法的引入大幅降低了计算资源与时间成本,并能与实验数据结合,进一步提升预测可靠性,甚至在实验数据存在噪声时仍能保持可靠性。
面临的挑战:
首要问题在于模型训练所需的高质量、相关性强且多样性充足的数据难以获取,且成本高昂,导致基于深度学习的工具在面对未见过蛋白复合物时预测能力有限。
另一项挑战在于模型的可解释性:深度学习通常难以提供其预测结果的理论依据,这在诸如药物发现等需高可信度的领域中可能影响其被接受程度。
此外,深度学习方法侧重于捕捉全局相互作用,而药物设计常关注特定位点,此时传统对接方法在某些场景下可能仍更具适用性。
深度学习模型也存在一些与传统模型一样的缺陷,例如,对于结构较大且非常复杂的蛋白质难以预测其结合模型,对于结构无序的蛋白质也难以预测其结合模型。
对于存在多个蛋白结合位点和能与多个蛋白相互作用的蛋白质,深度学习模型通常被训练预测一个结合位点。
尽管如此,深度学习方法近年来已经越来越受欢迎,超越了传统工具。它们可能彻底改变蛋白-蛋白对接领域,使准确预测蛋白-蛋白复合物成为可能。
AlphaFold-Multimer
AlphaFold是由DeepMind开发的革命性工具,它能够根据氨基酸序列预测蛋白质的三维结构。该工具的初始版本专注于单体蛋白质结构预测,而后续推出的AlphaFold-Multimer和AlphaFold3则进一步扩展了功能,能够高效预测蛋白质复合物的结构。
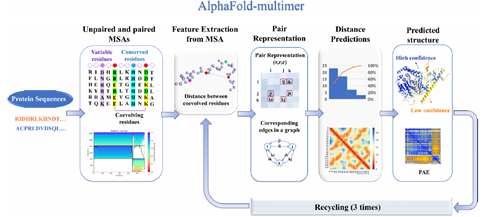
AlphaFold-Multimer预测蛋白复合物流程
AlphaFold-Multimer 利用同源序列搜索为目标序列构建多序列比对(MSAs),并可进一步从PDB数据库中选取结构模板。
通过对MSAs的分析,系统提取出反映进化关系的特征,包括保守残基和潜在相互作用位点。随后,模型生成残基对以推断氨基酸之间的相对位置,并借助概率分布距离预测残基间距离。此外,AlphaFold-Multimer 针对同源复合物的对称性进行了专门优化。
在模型评估方面,AlphaFold-Multimer 采用多种指标:每个原子的局部距离差异测试(pLDDT)得分(范围0-100)用于衡量预测结构的置信度;预测对齐误差(PAE)估计残基或结构域之间的预期位置偏差,以评估结构一致性;预测模板模型得分(pTM)反映整体结构的准确性;而界面预测模板模型得分(ipTM)则专门评估复合物界面残基间的预测准确性。
AlphaFold3在AlphaFold2和AlphaFold-Multimer 的基础上进一步发展,通过多项创新提升预测精度。其采用扩散模型架构直接生成原子坐标,避免了传统基于预定义表示方法的限制。
同时,AlphaFold3 以 Pairformer 模块替代 Evoformer,显著提高计算效率,降低对多序列比对的依赖,并能够更有效地学习残基间相互作用,拓展了在各类生物体系中的应用范围。
06 蛋白-蛋白对接工具的评估
在使用基准蛋白数据库对蛋白质-蛋白质对接工具进行性能评估与比较的过程中,不同工具之间表现出显著差异:
根据CAPRI的可接受标准,在Benchmark 4.0上的评估结果显示,ZDOCK 3.0.2在最佳预测结果中的成功率为12%,而在前50个预测中的成功率达到51%。
SwarmDock每次运行最多可耗时36小时,但其报告的成功率与ZDOCK相近。
在从头对接任务中,HADDOCK的表现与ZDOCK相当,但在基于实验数据的对接中表现更优。
然而,其总体性能仍落后于一些表现一贯优异的工具,如ClusPro——该工具在CAPRI评估中持续领/先,显著优于HADDOCK、GRAMM-X和LzerD等竞争对手。
具体而言,ClusPro在2009年(41.67%)、2013年(42.86%)和2016年(62.28%)均取得了基于可接受及以上质量标准的预测成功率。
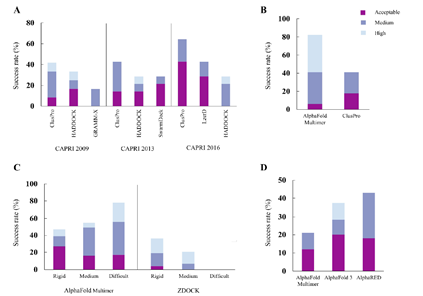
不同对接工具预测成功率比较
RosettaDock以其在预测近天然结构方面的高准确性著称,在未结合状态的局部对接中成功率超过60%。该工具在不同难度级别的对接任务中也表现出稳健性能:刚性对接成功率为58%,中等难度为30%,高难度案例为14%。在CAPRI第3–5轮中,RosettaDock以中高精度成功预测了全部五个中等及以上难度(残基数<450)的复合物结构;但在第6–12轮中表现有所回落,仅成功对接了2个靶标。
在Benchmark 5.0包含的230个中等至高质量蛋白质复合物上,PatchDock的成功率仅为2.61%,显著低于其他工具;而RosettaDock成功率最高,达52.61%,其后依次为FRODOCK(20%)、ZDOCK(16.09%)和ATTRACT(10.43%)。
在对19个靶标复合物进行运行时间测试时,ZDOCK耗时最短(6分钟),PatchDock、ATTRACT和FRODOCK约为15分钟,而RosettaDock耗时最长(230分钟)。
类似AlphaFold的深度学习方法极大推动了蛋白质对接领域的发展。在Benchmark 2包含的17个异源二聚体上,AlphaFold-Multimer基于DockQ评分达到了82.34%的可接受及以上准确率,优于ClusPro的41.16%。
在DB5.5的152个测试案例中,其表现也更为出色,尤其在挑战性场景中优势明显:
AlphaFold-Multimer在可接受及以上准确率的模型中,刚性、中等和困难案例的成功率分别为47%、55%和78%,而ZDOCK则显著下降(36%、19%和0%)。
此外,AlphaFold3在蛋白质-蛋白质复合物预测中的成功率达到76.7%,优于AlphaFold-Multimer v2.3的67.5%;
在蛋白质-抗体复合物中,AlphaFold 3同样表现更优(DockQ > 0.23,基于1000个样本),成功率为63%,对比于AlphaFold-Multimer的29.5%。
共识方法通过整合传统策略与深度学习模型,进一步提高了对接准确性。
例如,AlphaRED在DB5.5基准数据集上成功率达到63%,优于AlphaFold-Multimer的54%;在SabDab数据集上,其成功率为43%,而AlphaFold 3和AlphaFold-Multimer分别为37.5%和21%。
尽管对接工具的成功率为功能比较和工具选择提供了有用参考,仍需考虑以下因素:
首先:不同研究对“成功”的定义不一,有些侧重精确结合姿态,有些则接受近天然构象;
其次:工具性能受数据集特性影响,如复合物性质(柔性/刚性)、规模与复杂程度;
第三:评估指标不一也增加了比较的复杂性,同一工具可能在不同指标中表现不一致;
第四:评分函数因工具而异,某些在特定复合物类型(如酶-底物、抗原-抗体)上表现更优;
第五:软件版本更新会持续影响性能,因此评估结果具时间敏感性;第六,部分工具参数设置复杂,需专家操作才能实现高精度,而全自动化工具则更易使用;
最后:依赖实验数据的工具其准确性受数据质量制约。
07 结论与工具选择指南
结论
蛋白质-蛋白质对接在药物设计和细胞生物学过程研究中具有核心作用。尽管目前已开发出多种预测蛋白质复合结构的工具,但每种工具均存在其独特的优势与局限性。
未来该领域的研究重点包括发展新算法以更好地处理构象采样、蛋白质柔性及结合位点复杂性,改进评分函数,优化力场参数,并结合实验数据推动方法学进步。
同时,引入深度学习、强化学习、集成对接及混合策略等先进技术,有望为理解蛋白质功能带来突破性进展。随着领域发展,深度学习与传统方法融合已成为趋势,并将推动更强大、更可靠对接工具的出现。
选择指南
一般而言,选择最适合的对接工具需综合考虑用户的具体需求、蛋白质特性以及可用计算资源。
→对于刚性无约束对接或对称多聚体,PatchDock、ZDOCK、ClusPro 和 SymmDock 较为高效。
→若需支持柔性对接或引入实验数据约束,HADDOCK、RosettaDock和SwarmDock 能显著提高预测准确性。
→针对抗体-抗原或酶-抑制剂复合物,推荐使用RosettaDock、DockGPT 和 DiffMaSIF 以获得较高精度。
→pyDockSAXS 和 FoXSDock 等工具能够整合SAXS数据,而 JabberDock 在膜蛋白对接方面表现优异。
→对于无序蛋白质,可选用 LzerD 和 IDPdock。
→若需实现自动化或大规模对接,Hex、SnapDock 和 ReplicaDock 是较有效的选择。
→在缺乏高质量结构数据的情况下,可选用 HDOCK、InterEvDock3 和 HADDOCK 等进行从头或模板引导的对接。
→AlphaFold 凭借其基于序列的高精度预测、对修饰残基、配体及多链复合物的良好支持,已成为一类代表性方法。
→PRODIGY 和 HawkDock 则在基于 MM/GBSA 的结合亲和力估计和模型排序方面具有重要价值。
因此,为实现更准确的对接预测,应在明确研究问题与约束条件的基础上,仔细评估各工具的优缺点,合理选用多种工具并比较其结果的一致性与可靠性。
Ref1:Barhoon M, Mahdiuni H. Exploring Protein-Protein Docking Tools