- 移动端


上海吉凯基因医学科技股份有限公司品牌商
15 年
手机商铺
- NaN
- 0.5
- 0.5
- 1.5
- 0.5





公司新闻/正文
虚拟筛选在药物发现中的应用
354 人阅读发布时间:2025-07-18 12:40
计算机虚拟筛选技术在早期药物发现过程中发挥越来越重要的作用,通过虚拟筛选大规模商业可及化合物库,快速获得命中分子,能够大幅度缩短小分子药物发现的周期,提高候选化合物发现效率并大幅度较低早期药物研发的成本。然而,虚拟筛选的假阳性分子比率过高是其明显的缺陷。例如,某AI制药公司公布其使用传统的虚拟筛选方法阳性分子命中率仅1-2%,通过优化虚拟筛选方法,阳性分子命中率提高到了26%,这是非常大的进步,但是仍然有70%以上的分子是非活性的。除了从软件入手改进筛选方法提高阳性率以外,还可以从其他方向改进虚拟筛选的阳性率。本文将介绍几种选择候选分子的方法,帮助大家提高虚拟筛选的命中率。
选择合适的骨架结构
药物化学家将骨架分为功能性骨架和结构骨架。功能性骨架包含分子中与靶点相互作用的所有因素的集合。药物化学家根据功能性骨架优化分子的活性、选择性和PK、PD性能。结构性骨架是一种在适当的几何形状中提供出口矢量的支架,以允许引入关键的相互作用部分来修饰骨架。化学信息学家使用一套非常有效的分子组成成分表示方法来概括分子骨架结构,包括环系统(ring system)、链接子(linker)、支链(side chain)和分子框架(framework)。
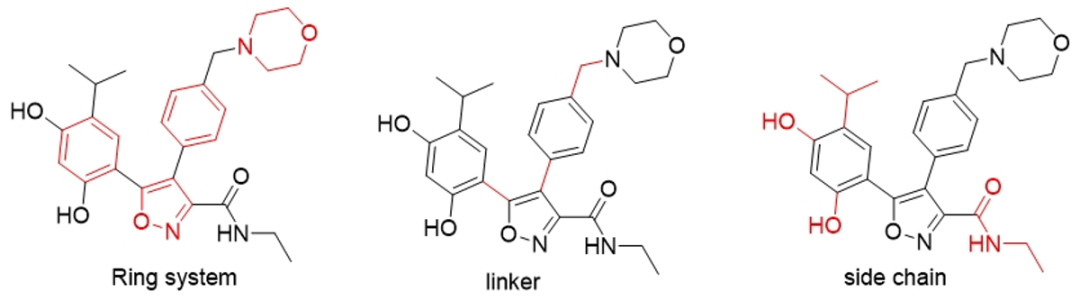
从单个分子生成分子骨架的方法是用分子框架(Murcko框架)和图形框架表示。分子框架是通过修剪所有不连接两个环状系统的非环状子结构,从单个分子生成骨架结构。图形框架是一种进一步的抽象,其中原子标签和和化学键被丢弃,以提供对一种拓扑结构的简单抽象。
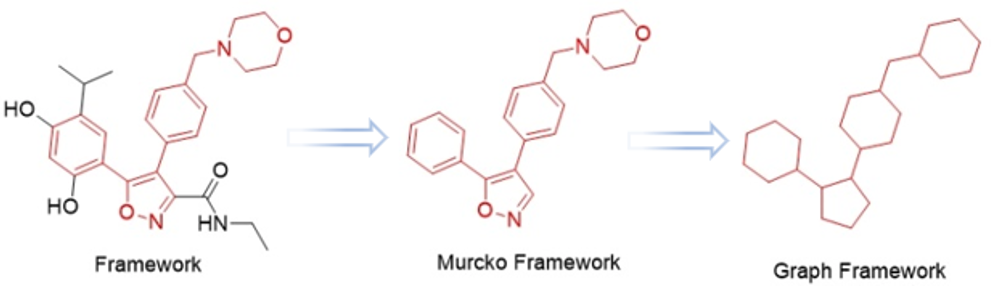
了解了分子框架,我们就能快速的将虚拟筛选的候选分子简化为一个个由环和linker连接的骨架结构。分子的骨架结构通常决定了分子与靶点的特异性结合,而特异性结合决定了分子的生物活性,包括治疗效果和毒性反应,分子的特异性结合通常也决定了靶点选择性。所以,选择合适的骨架结构对早期获得活性分子和后期从骨架结构出发进一步进行活性和选择性优化都非常重要。骨架架构通常选择由两个或者三个环状结构构成的核心结构,其结构通常是刚性的,而核心结构之外的结构可以看成是支链结构,这些支链结构在后期可以被取代或者删除来优化药物的活性和选择性,而核心结构被改变之后往往会失去对靶点的活性。
虚拟筛选可以通过计算化学结构相似性和聚类分析来分析不同的骨架结构,通常在相似的骨架结构中我们最多选择一两个最优的分子来进行测试,这样虚拟筛选的结果就能够探索更广阔的化学空间,来发现更优的核心骨架结构。
核心骨架结构的选择对药物发现项目的成败发挥着非常关键的作用,性能优异的核心骨架结构不仅具有良好的体内外药效和药代动力学性能,而且便于后期进行结构优化和SAR研究。例如,某制药公司发明了磷酸肽类stat6抑制剂,该类抑制剂含有磷酸基团,造成药物细胞渗透性差,后期虽然通过前药设计屏蔽磷酸基团,改善了细胞渗透性和代谢稳定性,但是仍然有许多问题未解决。至今,项目已经过去了20多年,该类抑制剂仍然处于临床前研究阶段。
使用重原子效率提高筛选命中率
在虚拟筛选过程中,通过打分函数对不同分子量的分子进行打分,通常分子量较高的分子的打分要优于分子量低的分子打分,两者是正相关的。例如,下图中某靶点虚拟筛选结果中打分值S2(截断值S2=-7.7)和分子量的关系图,从图中可以看出,分子量低于400的分子打分值低于-7.7的分子数量很少,而打分值较优的分子基本上分布在400-470之间(考虑类药性,筛选分子量在250-500之间)。如果虚拟筛选的命中分子按照打分值排序进行选择,那么分子量低的分子就完全没有机会被选中,这样就失去了选择较优骨架结构分子的机会。
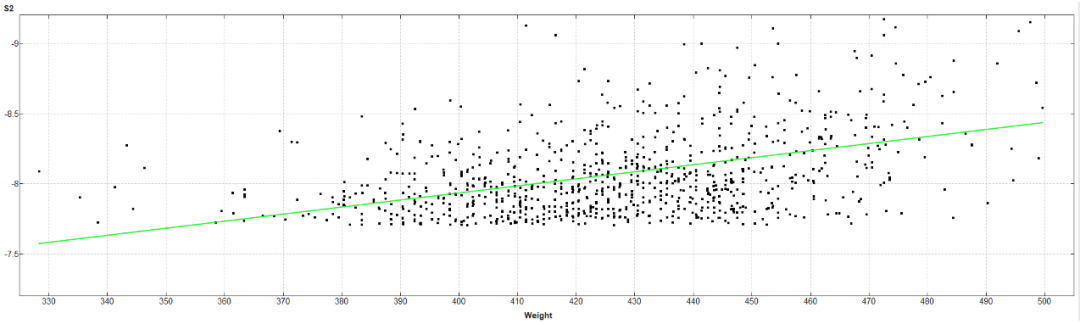
事实上,分子量较大的分子虽然打分较高,其活性并不一定比分子量低的化合物要好,而且由于其分子量较大,其溶解性和渗透性不如低分子量的化合物,在体内外药效验证实验中会带来不必要的麻烦。另外,分子量大的分子很可能包含对药效作用不必要的取代基团,后期要花费大量的时间和精力进行SAR研究,优化掉这些不必要的基团。
为了在虚拟筛选中兼顾分子量和打分值,使用配体效率 (Ligand Efficiency, LE)这一参数来综合评价候选分子。配体效率是一种根据分子中每个原子与靶点结合的平均结合能量来比较分子的方法。LE的计算公式为LE=ΔG/HA, ΔG是分子与靶标的结合自由能,HA是分子的重原子数量,在虚拟筛选过程中ΔG可以用分子对接打分值S代替。从已报道的药物统计数据分析,LE可接受的值为LE>0.3kcal/mol/HA。低于这个数值的分子代表其成药性较差,后期可能需要大量的优化工作来提高活性和成药性。在虚拟筛选过程中,LE的数值范围可以放宽一些,例如,有些靶点由于其结合口袋较小造成所有分子的打分值都不太理想,此时可适当放宽LE值至0.25kcal/mol/HA,再在满足LE和S值条件的分子列表中挑选骨架较优的分子。
可旋转键数量的考虑
可旋转键是指药物分子中的非末端单键,它允许键两端连接的基团自由旋转,常见的可旋转键包括碳碳单键、碳氧单键、碳氮单键等。分子中包含大量的可旋转键,会造成分子的可能构象增加,在通过生物膜时,分子从无序构象转变到有序状态,会造成系统的熵损失,这在热力学上是不利的,从而降低了膜通透性。可旋转键数目较多与口服利用度低相关,在libinski类药5原则中规定,药物的可旋转键数量不超过10,满足类药5原则的的药物分子可能具有较好的口服生物利用度。其次,高度灵活的分子可能更容易暴露出易被代谢酶(如细胞色素P450)攻击的位点,构象变化可能使原本不易被攻击的位点暴露出来,从而增加被代谢失活的风险,降低代谢稳定性。再次,高度柔性的分子虽然可能有某个构象非常适合结合靶标分子,但是在溶液中该构象的占比可能很低,需要靶蛋白通过“构象选择”机制来捕获这个构象,这可能影响药物的结合动力学。最后,高灵活性的分子由于其具有多个构象,其特异性可能较低。例如,其某个构象可能结合其他靶标分子,造成脱靶效应。
在药物虚拟筛选过程中,通过设定阈值来限制可旋转键的数量,比如,按照类药5原则设定rotN小于10。在10个可旋转键以内还可以通过以下方法来过滤候选分子,比如,在核心骨架结构之间,可旋转键(连接不同环结构的linker)最好是1-2个,核心骨架结构保持较好的刚性结构。如果多个可旋转键连接一起,分子的刚性将大大降低,这样的结构最好出现在不太重要的侧链上以保持核心结构的刚性。有些基团由于其旋转受限,可以看成是刚性结构,比如常见的有酰胺基团和磺酰胺基团,这些基团可看作是核心骨架成分。
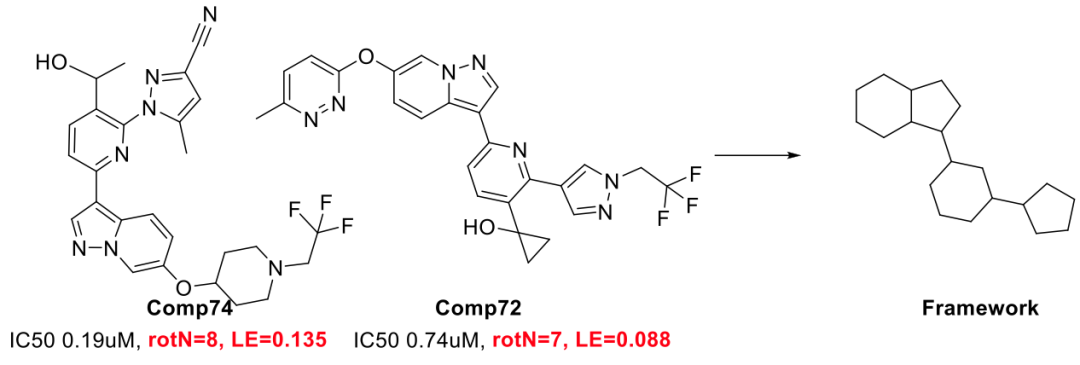
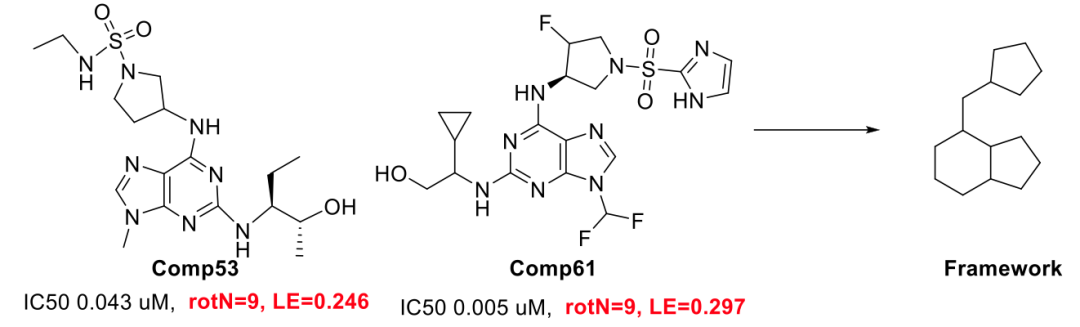
最后,我们一起来看一下2025年几个小分子药物专利公布的骨架结构、LE和rotN数据:
AHR激动剂,礼来公司,WO20250920758A1
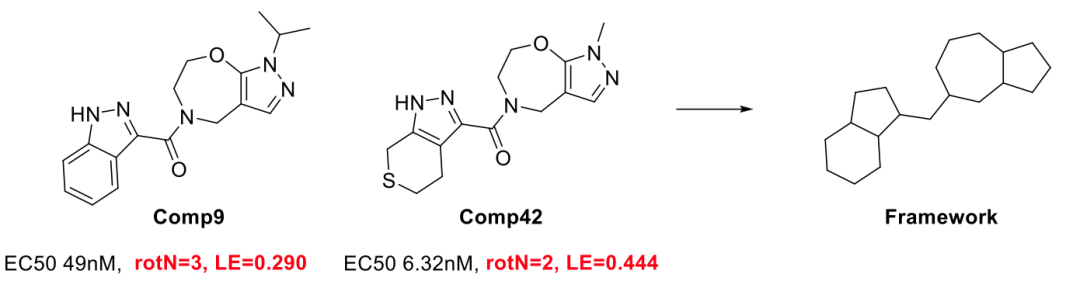
SIKs抑制剂,罗氏制药,WO2025017078A1
CDK2抑制剂,阿斯利康,WO2024127350A1
AVPR1A拮抗剂,迈阿密大学,WO2024197124A2